FAQ – Frequently asked questions about MG-RAST¶
The answers to some of these Frequently Asked Questions can be found elsewhere in this manual, they are listed here for users who would like a quick answer to a simple question. Other sections of the manual will generally contain more detail than the answers in this chapter. Some answers are just links to relevant sections in other chapters.
General¶
What is MG-RAST?¶
The MG-RAST server is an open source system for annotation and comparative analysis of metagenomes. Users can upload raw sequence data in fasta format; the sequences will be normalized and processed and summaries automatically generated. The server provides several methods to access the different data types, including phylogenetic and metabolic reconstructions, and the ability to compare the metabolism and annotations of one or more metagenomes and genomes. In addition, the server offers a comprehensive search capability. Access to the data is password protected, and all data generated by the automated pipeline is available for download in a variety of common formats.
Contacting the MG-RAST team and help desk¶
The MG-RAST project uses a ticket system to manage interactions with users, please use the email address for the MG-RAST project shown in Figure 9.1.
The email address for the MG-RAST project. Note that it was inserted into this document as an image and can not be copied as text, you will have to type it.
We recommend including as much detail as possible into your emails to the help-desk, details like account names, MG-RAST identifiers will help us identify any issues and speed up resolving them.
Below are examples of the types of details we would like to receive:
- your name
- your account name for MG-RAST (please do NOT include your password or webkey)
- a clear text description of your problem
- any MG-RAST identifiers (those are the 444xxxx.3 numbers)
- any project numbers
- the browser and which version you are using, if the problem relates to the web site
- what platform your data was created on
- if your data was a failure in the web site, what time the failure occurred
- the URL and name of the page you were viewing
- screenshot(s) of the problem
What kinds of data sets does MG-RAST analyze?¶
MG-RAST is designed to annotate a large set of nucleotide sequences, not a complete genome and not amino acid sequences. The RAST server should be used if you want to annotate complete, or nearly complete prokaryotic genomes. Version 3.2 accepts reads of length 75bp and up, and is capable of handling sequences of several dozen kilobases. For whole metagenome shotgun data we use a gene prediction step that is not suitable for eukaryotes, for that reason do not expect MG-RAST v3.2 to work with eukaryotic data sets or for the eukaryotic subsets of your data.
How many metagenomes can I submit?¶
We do not restrict user submission of samples. However, the computation required is massive and samples are processed on a first-come, first-serve basis. MG-RAST v3 is over 200 times faster than the previous version. We will also provide a CLOUD client (shortly after the initial release) that connects to MG-RAST and will allow you to add processing power to your jobs in MG-RAST.
Can I use MG-RAST as a repository for my metagenomic data?¶
MG-RAST has become an unofficial repository for metagenomic data, providing a means to make your data public so that it is available for download and viewing of the analysis without registration, as well as a static link that you can use in publications. It also requires that you include experimental metadata about your sample when it is made public to increase the usefulness to the community. We undertake to maintain public datasets within MG-RAST and they are not subject to deletion.
Who should I contact with questions or problems with MG-RAST?¶
All questions, comments or problems regarding MG-RAST should be directed to our support team using either the letter symbol in the navigation toolbox or via email to:
help at mg-rast.org
.
How should I link to MG-RAST in a publication?¶
You can provide a stable link to an MG-RAST job or project using the following URLs:
http://mg-rast.org/linkin.cgi?metagenome=
http://mg-rast.org/linkin.cgi?project=
For example, for the metagenome ID 4440283.3 the URL is:
http://mg-rast.org/linkin.cgi?metagenome=4440283.3
This URL provides a stable method of linking to your data which does not require the viewer to have an MG-RAST account. Please do not use the URL you see when you are browsing the site.
Note that by default your data is not visible to others, you will need to explicitly grant permission for it to be visible to anyone on the internet by making it public through the MG-RAST website.
Identifiers¶
MG-RAST automatically assigns a unique identifier to every dataset
submitted. Upon completion of the automated pipeline, datasets can be
viewed via the web interface by using the identifiers. The dataset
identifiers are of the form integer prefix.revision. An
example is 4440283.3.
In addition to individual datasets, projects (groups of datasets) can be
addressed with simple numerical project identifiers. An example is
128.
Linking to MG-RAST¶
Because future versions of MG-RAST may change, we provide a link-in
mechanism as a stable way of linking to MG-RAST. To link to datasets or
projects in MG-RAST, users should always use the linkin.cgi,
especially in publications.
Note: You must make the data set PUBLIC before you can publicly share the link. It will not work for others until you do.
Note: Do not use the URL that is displayed in the browser when browsing the site.
https://mg-rast.org/linkin.cgi?metagenome=
https://mg-rast.org/linkin.cgi?project=
For example, for the public dataset with metagenome ID 4440283.3 the
URL is: http://mg-rast.org/linkin.cgi?metagenome=4440283.3. For the
public project with project ID 128 the URL is:
http://mg-rast.org/linkin.cgi?project=128.
These URLs provides a stable method of linking to data that does not require the viewer to have an MG-RAST account.
Privacy¶
By default, a user’s data is not visible to others; the user needs to explicitly grant permission for the data to be visible to anyone on the Internet, either by sharing with individuals or by making it public through the MG-RAST website. Only the owner of a dataset (the original submitter) can make a dataset public and this requires explicit action on their part, MG-RAST does not make data public without this action. Owners can grant anonymous access to manuscript reviewers (see Section [section:reviewer_sharing]).
The web interface allows sharing and publication of data, requiring the presence of minimal metadata (see Section 4.7) for data that is made public. Data can be shared or made public only after the computation has finished.
Sharing with individual users¶
Data and analyses can be shared with individual users. To share data,
users simply enter their email address via clicking the Sharing link
on the Metagenome Overview page. The dialogue shown in Figure
9.2 will allow entering email addresses.
Dialogue showing the sharing mechanism. The mechanism requires a valid email address for the user with whom the data is to be shared. A list of users with access to the data is displayed at the bottom on the page.
Both individual jobs as well as entire projects containing one or more jobs can be shared using a similar mechanism from the Job Overview and Project pages respectively.
As shown in Figure 9.3, we tend to see dataset sharing between small groups of users.
Data sets shared in MG-RAST by users (orange dots), shown as connecting edges.
Anonymous sharing with reviewers¶
To grant manuscript reviewers access to a project while preserving their anonymity click on the ’Create Reviewer Access Token’ button on the project page. This button is visible only to the owner of a project by clicking on the ’Share Project’ link. It will generate a token that can be sent to the publisher to pass on to reviewers. When a reviewer receives the token from the publisher they need to use the included link to access MG-RAST. If necessary the reviewer will need to register for an account and their account will have anonymous access to the project. The number of reviewers who have accessed the project is displayed to the owner in the list of users the project is shared with, but the identity of the reviewers is not disclosed. The owner of the project can revoke the token at any time to disable access.
Publishing¶
MG-RAST provides a mechanism to make data and analyses publicly accessible. All sequence data, metadata, analyses, and analyses files for a dataset will be freely available for download once it is made public. Only the submitting user can make data public on MG-RAST and once this is done it can not be reversed. Metadata is mandatory for dataset publication (see Section 4.7).
The following checklist describes the process of making MG-RAST datasets and projects public:
Ownership of the datasets: To make a dataset public your account needs to be labelled as the owner in MG-RAST.
Ownership of the project: Your account should be the owner of the project as well, this is usually just the account that was used to create the project.
Metadata: MG-RAST requires that you enter metadata for the project, samples and libraries before it is made public to increase its utility to the community. This is done through a pre-formatted excel spreadsheet which you fill in with the necessary metadata. If you have already entered metadata, e.g. during submission, and want to make changes, you can download this file with the existing metadata prefilled from the project page with the ‘Export Metadata’ link.
If you have not entered metadata for your project, download the latest metadata template file from: ftp://ftp.mg-rast.org/data/misc/metadata/ The first sheet is a README containing some important tips for entering the metadata. The second row in each sheet in the template contains some explanation and instructions for each column. The columns marked with red headers are required.
You can enter your data directly into the template, a better route would be to use the tool we built to facilitate metadata entry – MetaZen: http://mg-rast.org/metazen.cgi. MetaZen will step you through the data entry and then give you a pre-filled excel spreadsheet to download which you can then edit further if necessary.
Once you have the metadata file ready, upload with the ‘Upload Metadata’ link on the project page.
Release metagenomes: Make each dataset public, there is a ‘Make public’ link in the blue bar near the top of the Metagenome Overview page.
Project Data: Edit the project page information if you wish with the ‘Edit Project Data’ link. You can enter an abstract, links to publications, additional description, contacts etc. This page is the central point in MG-RAST from where people will access your data and analyses so add all information that may be useful.
Final step: Make the project public from the project page (project page blue bar, ‘Make Public’).
The link for a public project which should be used in a publication is listed near the top of the project page, e.g.: http://mg-rast.org/metagenomics.cgi?page=MetagenomeProject&project=128 where 128 is the MG-RAST project ID.
The link for individual public metagenomes which should be used in a publication is listed near the top of the metagenome overview page, e.g.: http: //mg-rast.org/linkin.cgi?metagenome=4440283.3 where 4440283.3 is the MG-RAST metagenome ID.
The publication to cite for MG-RAST is at http://www.biomedcentral.com/1471-2105/9/386.
Is MG-RAST open source and can I install it locally?¶
MG-RAST is indeed open source. We make the current stable versions available on github: https://github.com/MG-RAST/ However MG-RAST is a complex system to install (note: we have not been funded to create a readily installable version) and even more complex to operate. We advise against attempting to create a private installation and can not provide any help installing MG-RAST locally.
If you are a biologist worried about runtime of your jobs, there is a way to run your jobs on computational resources provided by you that will significantly help. Please contact us at our usual address mg-rast at mg-rast.org to inquire about ways of setting this up.
If you are a bioinformatician and want to contribute code or test alternatives for individual steps, we are currently preparing a system that will make all components of MG-RAST easily accessible. This is not currently sea-worthy. Same as with the biologists, please contact us at help at mg-rast.org for details.
Accounts¶
The analyses of all public datasets in MG-RAST can be viewed in entirety without an MG-RAST account. An account is required to submit sequence data for analysis or view the analyses of datasets which have been shared with you.
Accounts are for individuals, not services or groups. In our experience account sharing (e.g. two or more users having access to the same username/password information) will always lead to problems, we strongly discourage account sharing.
As scientist typically will switch employers every few years we encourage users to provide two email addresses, the primary email address could be your work email, the secondary your private email. By providing a second email address you can avoid losing access to your account if and when you switch employers and your work email is no longer available.
Account registration¶
Use the “Register” link on the front page of the website to request an account with MG-RAST, you will need to enter a unique login name and email address along with other minimal information. Use an email address you use regularly as it will be used to communicate with you when necessary. After registering you will receive an automated email with a temporary password after your account has been authorized, usually within a day.
If you forget your password you can request a new password on the MG-RAST website using your login and registered email address, a new password will be generated and sent by email to this address.
Account webkey¶
The webkey is a unique string of text, e.g. “b8Dvg2d5DCp7KsWKBPzY2GS4i” associated with your account which is used by MG-RAST for identification purposes. Your webkey is valid for a limited time period after which it expires and will not work anymore. You can generate a new webkey at any time, even if your current webkey has not expired.
The MG-RAST website provides two locations where you can generate a new webkey:
- Log in to MG-RAST and go to the Account Management page. Press the button under “Preferences” to go the the Manage Preferences page where the Web Services section displays your current webkey with its termination date. Click on the “generate new key” button to generate a new key and then click the “set preferences” button.
- Log in to MG-RAST and go to the Upload page and click on the “generate webkey” button in the “upload files” tab and then click on the “generate new key” button.
Note that generating a new webkey will invalidate your old webkey and your new webkey will be valid until the termination date displayed on the page.
Why do I need to register for this service?¶
If you do not plan to submit data for analysis to MG-RAST and only want to browse data which is publicly available there is no need to register. Otherwise we request that users register, with a valid email address, so we can contact you once the computation is finished or in case user intervention is required.
I have forgotten my password, what should I do?¶
In the navigation toolbox (top right corner of the webpage) there is a ’Forgot?’ link displayed. Click on this and enter your login and the email address you registered with MG-RAST. A changed password will be sent by email to this address. For security purposes you should login and change this new password as soon as you receive the email.
Can I change my account information?¶
Yes, you can change or modify your password, email address, name and funding source for your account. Login and make the changes on the account management page.
Upload and Submission¶
MG-RAST was designed to allow users to upload sequence data directly from next-generation sequencing machines. Data can be in FASTA, FASTQ, or SFF format.
We suggest uploading raw data (in FASTQ or SFF format) and letting MG-RAST perform the quality control step because this approach will allow us to identify any issues with the sequencing run. Frequently, local quality control will identify some issues but mask others.
Compressing large files will reduce the upload time and the chances of a failed upload. Users can upload gzip (.gz) and bzip2 (.bz2) or Zip (.zip) files, as well as tar archives compressed with gzip (.tar.gz) or bzip2 (.tar.bz2).
It is not necessary to assemble data prior to upload to MG-RAST. The system has been optimized for short reads and can handle uploads of many hundreds of gigabytes.
Assembled data can be uploaded to MG-RAST and read abundance information for contigs can be imported as well from FASTA files. The “assembled” option for the pipeline will attempt to retrieve read abundance information from the FASTA sequence files.
Data submission via the web interface¶
To start uploading data to MG-RAST through the website, click on the green up arrow. Doing so opens the Upload page. On this page you can upload files, modify the files where needed, add metadata, and submit files for analysis.
The page has three stages (see Figure [fig:submission_stages]). The first “Upload” to upload, manipulate, and collect all the files required for a submission, and “Submit,” to create the MG-RAST job(s), set analysis parameters, and start the analysis. The last is “Progress”, where you can monitor your job status.
The flow for MG-RAST submissions via the web interface
[fig:submission_stages]
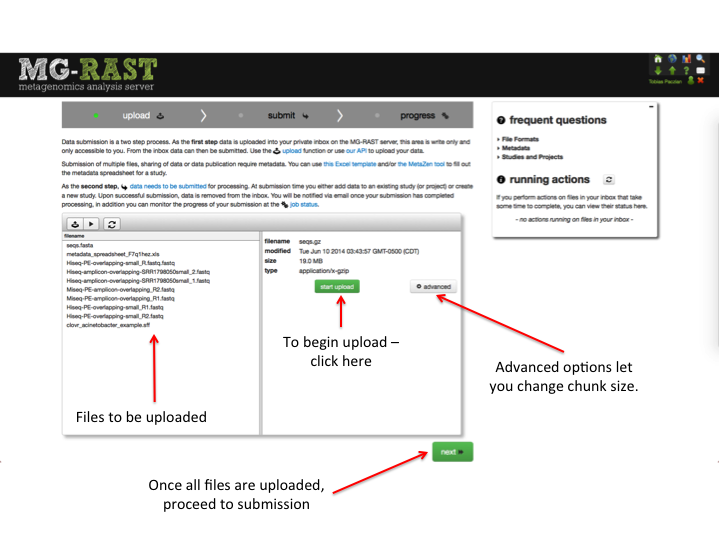
The MG-RAST upload page with its three main stages
[fig:upload_button]
Starting with version 3.6 of MG-RAST, the web upload page will provide significantly more user guidance and help with ensuring the files uploaded are both compliant with the required naming scheme and are transferred intact.
Data requirements for upload¶
Files larger than 50 MB should be compressed before upload, using gzip (preferable), bzip2 or Zip (less than 4 GB in size). Compression will reduce the time taken for the upload of the file, which in turn reduces the chance that the upload will fail. The requirements for submission are sequence information (required), metadata (strongly recommended) and barcode information (for multiplexed datasets only).
We note that priority will be giving to data that has compete GSC metadata and has been marked for eventual release to the public. The data release is under user control, MG-RAST staff will not release the data for the user.
To ensure files are uploaded properly, MG-RAST performs automatic MD5 [4]_ checking on client and server side (for most files) to ensure that files are received correctly by MG-RAST. This is an important part of data hygiene as files may get corrupted in flight. The new interface (from version 3.6 onwards), will check the integrity and will give you immediate feedback about whether your upload was successful. If not detected at upload time, a damaged file will lead to errors later in the pipeline, wasting both valuable compute cycles and, even more importantly, your time.
All files uploaded to MG-RAST should be named using only alphanumeric and ._ characters without spaces. As of version 3.6, the upload system ensures that files are compliant with the mandatory naming scheme, using only alphanumeric and .-characters without spaces. In addition, there is no need to extract/uncompress files after upload. MG-RAST does this automatically along with checking metadata and sequence file format and nomenclature compliance.
Advanced options provides the option to change chunk size. Chunked uploading allows us to break a large file into small chunks, and send these pieces to the upload server one-by-one. If an upload fails, we need only resume from the last successful chunk and allows for resuming uploads. As a rule, the larger the file and the faster your connection, the larger the chunk size should be. Set the size lower if your connection is slow. We have a default setting that works well for most data sets and connection speeds. If you are encountering upload failure (outside of formatting issues), try a smaller chunk size.
The following three kinds of files can be uploaded:
Sequence files
Sequence files must be in either FASTA, FASTQ, or SFF formats
Sequence file names must have one of the following extensions – ‘.fasta’, ‘.fna’, ‘.fastq’, ‘.fq’, or ‘.sff’.
FASTA and FASTQ files should be in plain text ASCII.
FASTA files (and all sequence data submitted to MG-RAST) should not contain protein sequences.
Assembled data with read abundance information must be in FASTA format and the coverage included in the sequence ID using the following simple format:
>sequence_number_1_[cov=2] CTAGCGCACATAGCATTCAGCGTAGCAGTCACTAGTACGTAGTACGTACC >sequence_number_2_[cov=4] ACGTAGCTCACTCCAGTAGCAGGTACGTCGAGAAGACGTCTAGTCATCAT ....
The abundance information must be appended without spaces to the end of the sequence name (also without whitespace) in the format “_[cov=n]”, where n is the coverage or abundance of each contig. Sequence files in this format should be submitted with the “assembled” option selected and the pipeline will retrieve read abundance information from the sequence file.
Metadata file
We provide a spreadsheet template that can be filled out with all the available metadata information for a dataset, there is a link to the template on the upload page. Download the template and edit to include as much information as is available. While the number of fields in the template is large, the number of required fields, colored in red in the template, is small. The template file can be used to upload metadata for one or multiple samples and submit them to MG-RAST as a single project. The metadata can be modified at any time after submission to add information or to correct errors. See Section [section:generating_metadata] for more details.
We note that a good strategy is to copy an existing metadata file and modify the values appropriately. Our experience has also shown that editing the metadata file with tools other than Microsoft Excel will corrupt the files.
Barcode file
Barcoding reads allows multiplexing multiple samples into a single sequence file. Barcode files allow demultiplexing those files. Consequently, Barcode files are required only for sequence data which will be demultiplexed on the MG-RAST website. In many cases (typically for shotgun metagenomes) the demultiplexing will have already been done by the sequencing center. If you have demultiplexed sequence data, you do not need to enter the barcodes associated with your samples in a Barcode file. While suitable for all kinds of barcodes and sequence data, we expect the built-in demultiplexing to be used mostly for custom barcoded amplicon sequences.
The barcode file should be in plain text ASCII.
If the sequencing facility generated the libraries and did not demultiplex them for you, make sure to get the barcodes corresponding to each of your samples. The barcode file should be in plain text ASCII, a downloadable example can be found at: ftp://ftp.mg-rast.org/data/manual/example/.
Each line of the file should contain a single barcode sequence followed by a tab and then a unique filename, with as many lines as necessary for the barcodes in the sequence file you are submitting. Additional columns are ignored.
Example: ACTCTCGTG sample_1 CAGACATCT sample_2 GTAGATCAC sample_3
The barcode file typically will be provided by whoever created the amplicons, in many cases that is the sequencing center.
Uploading data¶
In this first step, data is uploaded into your private inbox on the MG-RAST server, this area is write-only and only accessible to you. Data in the inbox cannot be read or re-exported, its sole purpose is to serve as a starting point for the pipeline.
When an upload is started it can be aborted or paused. Pausing will cause the current chunk to complete and then pause the upload. Abort will interrupt the upload immediately. A paused upload can be resumed by clicking the resume button in the upload dialog. Aborted uploads can be resumed or deleted just like other incomplete uploads by clicking the resume button in the top bar.
When an upload completes (that is not an archived file), an automatic md5 check will be calculated and the result presented to the user. In the case of an archive file uploaded, the user has to produce the checksum of the local file themselves and can paste it into a check field for validation. A note will be displayed to the user to calculate the md5sum on the uncompressed file. Archived files will be decompressed automatically.
At upload:
- Sequence files will automatically trigger sequence stats calculation
- Sequence files with calculated stats will display those stats upon selection
- Sequence files will show buttons for demultiplexing and joining of paired ends
- Barcode files will automatically show a button for demultiplexing
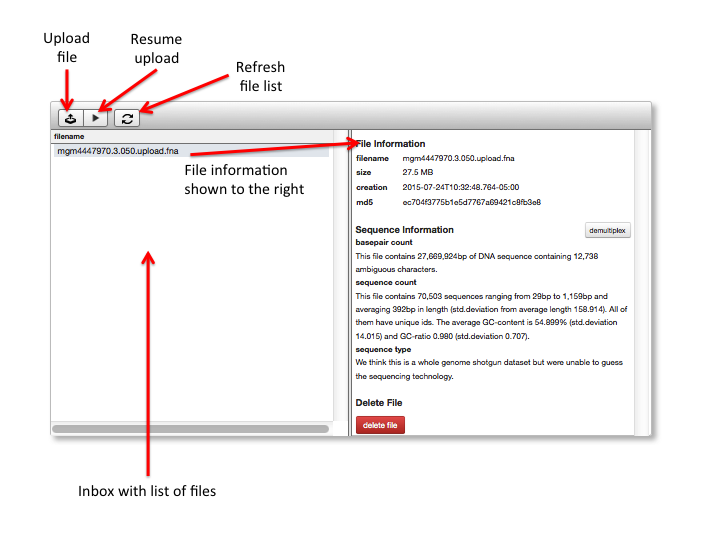
The main elements of the file browser explained. The left side pane shows a list of uploaded files. The top bar provides available actions. Users can select files to view information and whether the file passes formatting check.
[fig:upload_inbox]
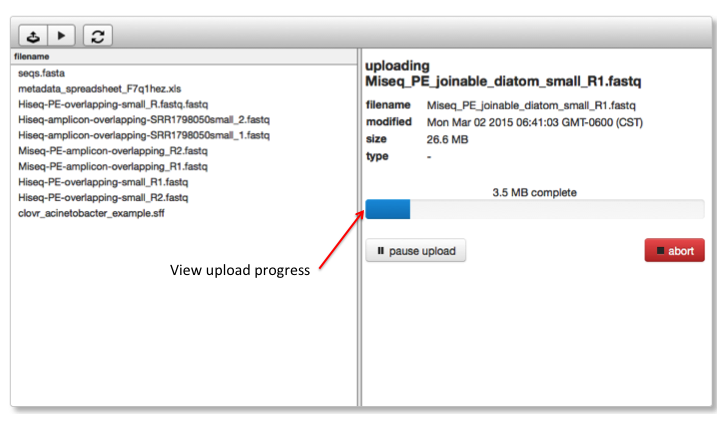
Once selected from the file browser you can start the upload and observe progress in the right side pane.
[fig:upload_progress]
From the inbox data needs to be submitted to the annotation pipeline. Once files are uploaded, the inbox allows a set of operations to be performed.
Expected upload speeds¶
Based on observed values, upload times per 1 GB (\(10^9\) bytes) vary from 2 minutes to over an hour, with typical times being 10 to 15 minutes. Your experience will vary depending on the speed of your connection to the internet and the quality of service in your region.
Table 1 summarizes observed upload times that might help users estimate how long the upload should take.
| Technology | Rate (bit/s) | Time for 1GB Upload |
|---|---|---|
| Modem 14.4 (2400 baud) | 14.4 kbit/s | 154 hours |
| ADSL Lite | 1.5 Mbit/s | 1.5 hours |
| Ethernet | 10 Mbit/s | 13.33 minutes |
| T3 | 44.736 Mbit/s | 3 minutes |
| Fast Ethernet | 100 Mbit/s | 1.33 minutes |
Frequent issues with data uploading¶
- Old browser version will not provide good throughput with the upload and may fail to execute the Javascript for the uploader properly. Update to the latest version of Firefox for optimal performance.
- Browser-add-ons have in several occasions blocked uploads or led to aborted uploads in the past. Disable those add-ons temporarily for the duration of the upload.
- In rare cases network devices have been presenting problems for the upload. Some institutions have not anticipated the use of the http protocol to transfer large data sets. In these cases the best option is to find another network location for the transfer.
File filters in place for uploaded files.¶
Since MG-RAST has been designed to work with metagenomic and metatranscriptomic datasets, there is a filter in place trying to identify datasets not suitable for MG-RAST. Those datasets will be colored red in the inbox listing and cannot be submitted. Following are the criteria for rejection:
- Protein sequences – MG-RAST is optimized to perform translation from DNA to proteins.
- Reads shorter than 75 basepairs – The gene prediction stage performance deteriorates significantly with shorter reads.
- genomes – Submissions with complete genomes or a small number of contigs are rejected as well. Here our sister service RAST at http://rast.nmpdr.org should be used instead of MG-RAST.
- Files that are too small (sequence data less than 1 Mbp) – Files that are too small for MG-RAST to properly function are rejected at the submission stage. The minimal size requirement is 1 megabasepair.
- Corrupted files – FASTA and FASTQ files which do not conform to the format standard, e.g. if the number of unique identifiers does not match the number of sequence records in a file, the file is considered corrupt.
- Alignments – We cannot identify proteins from sequences containing alignment information.
- Colorspace – The tool chain does not function for ABIsolid sequences in colorspace. Please translate to standard FASTA.
- rar compressed files and Zip files over 4 GB – We cannot decompress these files.
In addition we will filter at the upload stage any Word documents, Rich Text Format files, and all files without the extension .fna, .fasta, .fq, .fastq, or .sff in their name.
Note: We recommend computing an MD5 checksum and verifying that the checksum computed by MG-RAST is identical to the locally computed checksum. This is the best way to ensure data integrity.
Please note: After the actual upload is complete, the system will compute the statistics shown in Figure [fig:upload_inbox]. Computing this information takes some time, so the statistics for your sequence files will not be visible immediately after you uploaded it. If the statistics are not displayed in a reasonable time refresh your browser page to trigger the statistics computation.
Submit data for processing¶
In the second step, data needs to be submitted for processing. At submission time you either add data to an existing study (or project) or create a new study. Upon successful submission, data is removed from the inbox. You will be notified via email once your submission has completed processing. In addition you can monitor the progress of your submission at the job status.
- All submitted data will stay private until the owner makes it public or shares it with another user.
- Providing metadata is required to make your data public and will increase your priority in the queue.
- The sooner you choose to make your data public, the higher your priority in the queue will be.
The submission step provides a visual aid to identify completed tasks (the bars on the page are turning from blue (open) to green (done), see Figures [fig:submission_open] and [fig:submission_done]).
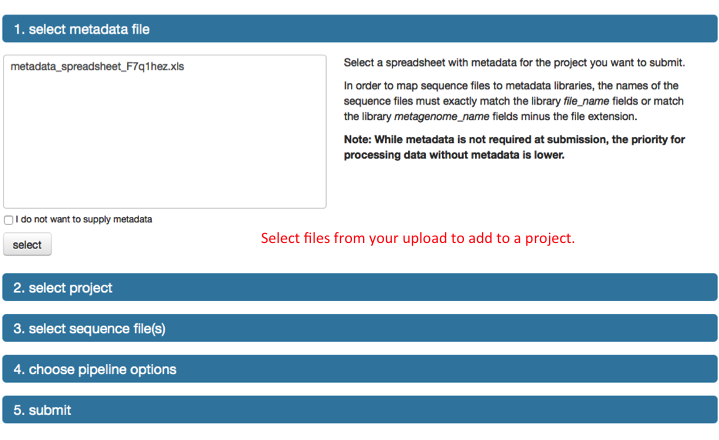
The submit page with none of the fields filled out.
[fig:submission_open]
The submit page with all bars in green indicating that the respective sections have been filled out.
[fig:submission_done]
Progress monitoring¶
Once data is submitted, you can monitor its progress.

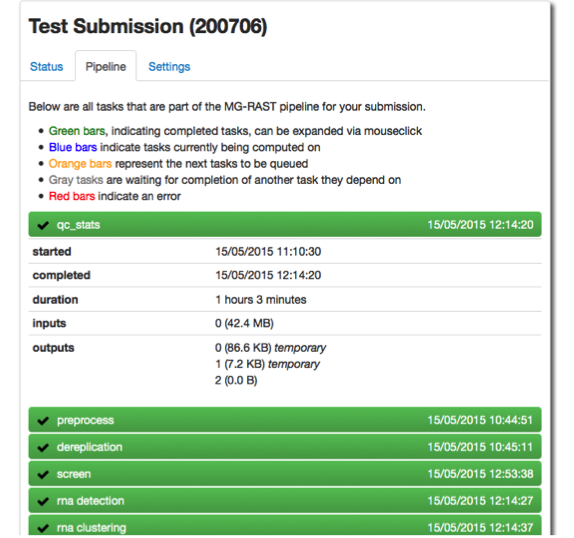
The jobs you have submitted are listed with their current status. A green dot indicates the stage has completed successfully, blue indicates that the current stage is in progress. Queued stages will produce an orange dot, green indicates a completed stage and red indicates error state. Gray dots will show for all stages waiting for other stages to complete.
[fig:submission_pipeline_view]
Depending on your priority (assigned based on available metadata and how public your data is) your jobs will progress through the system. Jobs that fail due to technical reasons (component failure etc.) will be restarted by MG-RAST staff.
You will receive an email once a given data set has finished processing.
Cmd-line uploader¶
The following upload instructions are for all file types supported by MG-RAST.
The mg-inbox command line tool allows upload of sequence and metadata files and management of the user’s upload area, the inbox. In order to operate on the inbox the user has to authenticate with an MG-RAST token. The token can be retrieved from the “Account Management” –\(>\) “Manage personal preferences” –\(>\) “Web Services” –\(>\) “authentication key” page via MG-RAST Web site.
Make sure you have python installed on your system.
https://pip.pypa.io/en/latest/installing.html
Go to the directory where you have your files to upload.
Download the upload script
ftp://ftp.mg-rast.org/tools/upload/mg-inbox.py
Checkout the help options for “mg-inbox.py”. If you received an error message that you are missing certain python libraries, you will need to install them before you can run the script. To install python libraries use pip install \(<\)lib-name\(>\) to install the missing libraries. Note: The error message from running “python mg-inbox.py –help” will provide the “lib-name” you are missing. You may have a few libraries to install.
python mg-inbox.py --help
First you need to login. The login option takes a user token and writes a login file after successful login. For example:
python mg-inbox.py login --token <myToken> python mg-inbox.py view all
You can upload a file into your inbox with
python mg-inbox.py upload <path_to_file>/<file_name>
If you have a compressed file to upload, supports gzip or bzip2
python mg-inbox.py --gzip upload <path_to_file>/<gzip_file> python mg-inbox.py --bzip2 upload <path_to_file>/<bzip2_file>
If you have an archive file containing multiple files to upload, supports: .zip, .tar, .tar.gz, .tar.bz2
python mg-inbox.py upload-archive <path_to_file>/<archive_file>
You can examine the content of your inbox with
python mg-inbox.py view all
You can submit your sequence files from the Upload page on the MG-RAST web site (cmd-line option coming soon).
REST API uploader¶
The following upload instructions are for using the MG-RAST REST API with the curl program. In order to operate the API the user has to authenticate with an MG-RAST token. The token can be retrieved from the “Account Management” –\(>\) “Manage personal preferences” –\(>\) “Web Services” –\(>\) “authentication key” page via MG-RAST Web site.
We strongly suggest that you use the scripts we provide, instead of the native REST API.
You can upload a file into your inbox with
curl -X POST -H "auth: <myToken>" -F "upload=@<path_to_file>/<file_name>" "https://api.mg-rast.org/inbox"
If you have a compressed file to upload, supports gzip or bzip2
curl -X POST -H "auth: <myToken>" -F "upload=@<path_to_file>/<gzip_file>" -F "compression=gzip" "https://api.mg-rast.org/inbox" curl -X POST -H "auth: <myToken>" -F "upload=@<path_to_file>/<gzip_file>" -F "compression=bzip2" "https://api.mg-rast.org/inbox"
If you have an archive file containing multiple files to upload do the following two steps, supports: .zip, .tar, .tar.gz, .tar.bz2
1. curl -X POST -H "auth: <myToken>" -F "upload=@<path_to_file>/<archive_file>" "https://api.mg-rast.org/inbox" 2. curl -X POST -H "auth: <myToken>" -F "format=<one of: zip, tar, tar.gz, tar.bz2>" "https://api.mg-rast.org/inbox/unpack/<uploaded_file_id>"